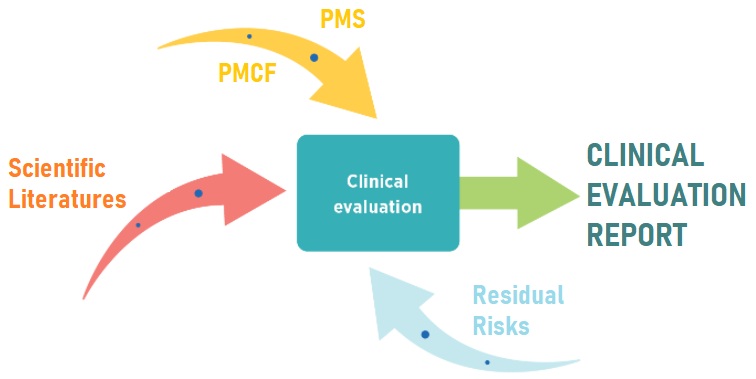
We prepared a final clinical evaluation report with examples about a medical device’s general safety and performance requirements based on the clinical data during the pre-market and post-market stages.
Identification, collection, appraisal, and analysis of the medical device’s pre-market and post market clinical data in order to demonstrate MDR GSPR compliance
- A clinical evaluation report template should be compiled to document the clinical evaluation with a specified time schedule.
- It should provide enough detail to understand the search criteria adopted, the available data, all assumptions made, and all conclusions reached.
- The contents of the CER are relevant to the medical devices, which should vary according to the nature and history of the device under evaluation.
A description of the device under the clinical evaluation plan should be provided. The scope of the clinical evaluation should include the identification of devices covered by this report, products, models, sizes, software versions, accessories, whether they are on the market, since when, in what regions, history of the device, and, in case of an update in the plan, including the date of past modifications with reasons and descriptions, sales volumes, and changes since the last report. The identification of the sections of the CER that are concerned with the new information and have been modified should be detailed.
We Provide Clinical Evaluation Report including:
MDR Clinical Evaluation Documentation
Here is an overview of how Clinical Evaluation Documentation can be done by MDR:
- Define the Scope of the medical device: The clinical evaluation plan should clearly define the device’s scope, intended use, patient population, and clinical indications. It should outline the methodology, conclusion, and documentation of the evaluation process, as well as provide clinical data certifying device compliance with safety and performance requirements.
- Collect Clinical Data: Collect clinical data from pre-market investigations, clinical trials, scientific literature, post-market surveillance, and adverse event reports. Document each data set, covering the device’s entire life cycle, for a comprehensive clinical evaluation report.
- Literature review: Perform a thorough review of the existing scientific literature to identify and analyze the relevant studies and reports. Consider clinical data related to similar devices or equivalent technologies. Describe the clinical background and identify the current knowledge or state of the art corresponding to the particular medical device. If equivalency is claimed, the validity of the criteria used for the demonstration of equivalence should be determined.
- Appraisal of pertinent data: The evaluators should evaluate each document individually in terms of its contribution to the assessment of the clinical performance and clinical safety of the device to ascertain the value of the data found in the literature search.
- Analysis of pertinent data: The analysis stage’s objective is to ascertain whether, when a medical device is used by its intended purpose, the appraised data sets available for the device collectively demonstrate compliance with each of the Essential Requirements about the clinical performance and clinical safety of the device.
- Risk-Benefit Analysis: Perform a risk assessment to identify potential hazards and risks associated with the device. Describe the measures taken to mitigate, control, or eliminate these risks. It also helps to identify the potential risk associated with the device as well as identify the benefits of using the device and make sure whether the benefits outweigh those risks.
- Clinical Evaluation Report (CER): A CER is an important technical document in the technical file which summarizes the clinical evaluation of a medical device. A clinical evaluation report should document the evaluation and its output, providing detailed search criteria, available data, assumptions, and conclusions. Create a Clinical Evaluation Report by compiling the clinical evaluation’s findings (CER). A Clinical Evaluation Report (CER) is a comprehensive, well-documented, and structured appraisal of clinical evidence to assess the safety and performance of a medical device. The CER should be kept up-to-date and reviewed at regular intervals.
- Consult with experts: To ensure the thoroughness and accuracy of the evaluation, seek input from qualified experts such as clinical specialists, statisticians, and regulatory affairs professionals. We must include the CV and declaration of interest of clinical evaluators along with the clinical evaluation report.
- Submit to Regulatory Authorities: In compliance with the guidelines and regulations of the appropriate regulatory body, prepare and submit the Clinical Evaluation Report. To approve or maintain the device’s market presence, the notified body will examine the report.
- Post-Market Surveillance: After the product is put on the market, keep an ongoing eye on its functionality and safety. If new information becomes available, update the clinical evaluation and note any adverse events or problems with the product.
- Periodic Safety Update Report: Provide regular updates, particularly for high-risk devices to regulatory bodies. Also, ensure compliance with ongoing reporting requirements.
Clinical Evaluation Documentation procedures vary by regulatory framework, so manufacturers must follow EMA or FDA guidelines and stay updated with regulations to maintain compliance with the European Medicines Agency (EMA) and US FDA. The demonstration of conformity with Essential Requirements, based on performance evaluation, bench testing, and pre-clinical evaluation, must be substantiated, and presented in a clinical evaluation report.
Benefits of appointing I3CGLOBAL for Clinical Evaluation Documentation
The following are the major advantages of appointing consultants and clinical writers for Clinical evaluation documentation as per current MDR
Active Post-Market Oversight: Active post-market oversight is crucial to ensure the safety and efficacy of medical devices. This involves continuous monitoring of devices after they have been approved and are in use. Manufacturers should actively collect data on device performance and report any adverse events. Regulatory authorities can use this data to identify potential safety issues, make necessary changes to device labelling, and, in some cases, recall devices if they pose a significant risk to patients. Active post-market surveillance can enhance consumer trust by demonstrating a commitment to safety and responsiveness.
High-Quality Clinical Evaluation Reports (CERs): Clinical Evaluation Reports are essential for demonstrating the safety and performance of medical devices. High-quality CERs are important to avoid regulatory rejections and to ensure that devices remain on the market. They should provide a comprehensive and transparent overview of the available clinical evidence and how it supports the device’s safety and efficacy. Meeting the required standards and guidelines for CERs is crucial to gaining notified body approvals and maintaining a strong regulatory standing.
Thorough Literature Search Strategy: A thorough, objective, and reproducible literature search strategy is vital for gathering the necessary data and evidence to support a medical device’s safety and efficacy. This strategy should be well-documented and should consider various reputable sources, including peer-reviewed journals, clinical trials, and relevant databases. It should also employ systematic review methodologies to ensure the quality and reliability of the gathered data. A well-executed search strategy is crucial in the scoping stage of CER development.
Faster Turnaround Time for CER Updates: Speed in updating CERs is essential, especially when new data or safety concerns arise. Rapid updates ensure that the clinical evidence for a medical device remains up-to-date and accurately reflects its performance. Delays in updating CERs could lead to regulatory issues or a loss of consumer trust. To expedite CER updates, it’s important to have efficient processes in place, maintain clear documentation, and stay informed about the latest clinical findings and regulatory requirements.
We have an in-house department which takes up post-market Clinical Follow-up, Retrospective Study, and Clinical Investigation.
In summary, a combination of active post-market oversight, high-quality CERs, a well-planned literature search strategy, and efficient CER update processes can help ensure the safety and efficacy of medical devices and also maintain Notified Body CE Certificate validity. Manufacturers need to stay vigilant, responsive, and proactive in managing their devices throughout their lifecycle.
FAQ's
What is Device Classification?
Classification of the device as per Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices should be mentioned.
What is know as Intended purpose
Exact description of the intended purpose as described in the device's IFU with exact medical indications (if applicable); claims made in available promotional materials. This section summarizes:
• Disease and its severity or aspects
• Medical indications
• Target population and intended user
• Body parts affected
• Duration of use
• Type of use (single or reusable)
What are to be considered under Device Description
• Device name
• GMDN code
• Brand name, models and variants, size and UDI-ID number
• Software versions and product code
• Physical and chemical description, including materials.
• Constructional materials
What is know by Data generated and held by manufacturer?
This section shall include details of clinical investigations, if any, PMS, PMCF, complaints, details of field safety corrective action, other user reports, relevant preclinical study data (bench test reports, verification and validation data).
What is the relevance of Bench Testing?
The results obtained from any external laboratory or in house and its evaluation in significance with clinical evaluation should be provided.
What are to be considered for next year evaluation
• The significant risks of device if high-risk or medium, high-risk target populations, severity of disease/ treatment challenges, duration of risk.
• Whether the device is well established or the innovation is there, the current level of confidence in the evaluation of clinical performance and clinical safety of the device from the data available from clinical investigations, PMCF studies, registries or other systematic studies, reports of risk or adverse event rates under the vigilance system.
• The clinical evaluation is actively updated with :
When new information is received from PMS that has the potential to change the current evaluation; if no such information is received, then at least annually if the device carries significant risks or is not yet well established; or every 2 to 5 years.
If the device is not expected to carry significant risks and is well established, a justification should be provided.